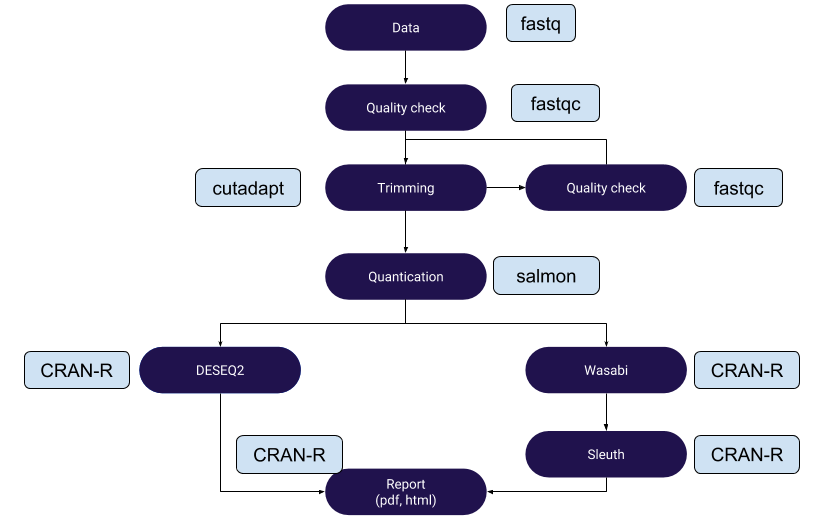
This repository has snakemake scripts for salmon-deseq2 pipeline for RNAseq data analysis



RNAseq data analysis by Salmon-DESeq2 and Salmon-Wasabi-Sleuth pipeline
RNAseq data analysis by Salmon-DESeq2 and Salmon-Wasabi-Sleuth pipelines are available here. RNAseq Data is availble from
Griffith Lab
. The workflow uses snakemake library.
Code Snippets
16 17 18 | shell:""" cutadapt --quiet -j {threads} -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT -o {output.R1} -p {output.R2} {input.R1} {input.R2} &> {log} """ |
6 7 | script: '../scripts/salmon_deseq2.R' |
13 14 15 | shell:""" fastqc -t {threads} {input} -q -f fastq -o results/cutadapt/ &> {log} """ |
13 14 15 | shell:""" fastqc -t {threads} {input} -q -f fastq -o results/fastqc/ &> {log} """ |
11 12 13 | shell:""" salmon index -p {threads} -t {input} -i {output.directory} --type quasi -k 31 &> {log} """ |
8 9 10 | shell:""" multiqc results -s -i "Project A results" -n "project A" -b "Salmon-DESEQ2/WASABI-SLEUTH results" -o results/multiqc -ip -q --no-data-dir &> {log} """ |
16 17 18 | shell:""" salmon quant -i {input.index} -l A -1 {input.R1} -2 {input.R2} -o {output.B1} -q --useVBOpt --gcBias --seqBias --posBias -p {threads} --numBootstraps 30 &> {log} """ |
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 | suppressMessages(library(vsn)) suppressMessages(library(tximport)) suppressMessages(library(readr)) suppressMessages(library(stringr)) suppressMessages(library(assertr)) suppressMessages(library(DESeq2)) suppressMessages(library(ggplot2)) suppressMessages(library(wasabi)) suppressMessages(library(apeglm)) suppressMessages(library(sleuth)) suppressMessages(library(pheatmap)) suppressMessages(library(regionReport)) # Store DESeq2 results dir.create('results/salmon_deseq2_results', showWarnings = FALSE, recursive = TRUE) # Load sample information samples=data.frame(samples=col_concat(str_split_fixed(list.files("./results/salmon"),"_",5)[,2:3],"_"), condition=str_split_fixed(list.files("./results/salmon"),"_",5)[,2]) row.names(samples)=samples[,1] ## Deseq2 workflow files=file.path("results/salmon",list.files("results/salmon"),"quant.sf") tx2gene=read.csv("reference/t2gene.dedup.tsv", sep = "\t",stringsAsFactors = F, header=F) salmon_data <- tximport(files, type="salmon", tx2gene=tx2gene) ddsTxi <- DESeqDataSetFromTximport(salmon_data, colData = samples , design = ~ condition) ## Filter transcripts with less than 10 counts keep <- rowSums(counts(ddsTxi)) >= 10 dds <- ddsTxi[keep,] dds # To be sure, make normal as reference condition dds$condition <- relevel(dds$condition, ref = "normal") dds # DESeq on DESeq2 object ddds <- DESeq(dds) # Extract results for comparison res <- results(ddds, coef="condition_tumor_vs_normal") # sort the results resOrdered <- res[order(res$padj),] ## Write results to Hard disk write.csv(as.data.frame(resOrdered),file="results/salmon_deseq2_results/condition_treated_results.csv") # Shrink the log values reslfc=lfcShrink(ddds, coef="condition_tumor_vs_normal", type="apeglm") # store the pics in pdf pdf(file = "results/salmon_deseq2_results/salmon_deseq2_results.pdf") # Plot counts for gene with lowest fold change (down regulated gene) plotCounts(ddds, gene=which.min(res$log2FoldChange), intgroup="condition") # Plot counts for gene with lowest adjusted p-value (statistically significant gene) plotCounts(ddds, gene=which.min(res$padj), intgroup="condition") # PCA for samples plotPCA(rlog(ddds), intgroup="condition")+theme_bw() # Distance plot for samples sampleDists <- as.matrix(dist(t(assay(rlog(ddds))))) cols=colorRampPalette( c("green","yellow","red"))(255) pheatmap(sampleDists, col=cols) ## Expression heatmap select=row.names(res[order(-res$log2FoldChange),])[1:20] cols=colorRampPalette( c("green","yellow","red"))(255) pheatmap(assay(rlog(ddds))[select,], col=cols) #Plot data post transformation (rlog) meanSdPlot(assay(rlog(ddds))) # Maplot for res plotMA(res) # Close the graphics device dev.off() # Generate report in pdf report <- DESeq2Report(ddds, project = 'Salmon-DESEQ2 workflow', intgroup = c('condition'), outdir = 'results/salmon_deseq2_results', output = 'index', theme = theme_bw(), browse = F,device = "pdf", output_format = 'pdf_document') ## Generate report in html report <- DESeq2Report(ddds, project = 'Salmon-DESEQ2 workflow', intgroup = c('condition'), outdir = 'results/salmon_deseq2_results', output = 'index', theme = theme_bw(), browse = F) ## Save the workspace save.image("results/salmon_deseq2_results/salmon_results.Rdata") ## Load the workspace # load("results/salmon_deseq2_results/salmon_results.Rdata") ## Wasabi and Sleuth workflow dir.create("results/sleuth_results") # # Wasabi workflow sfdirs <- file.path("results/salmon", c(list.files("results/salmon"))) sfdirs prepare_fish_for_sleuth(sfdirs) ## Preparation for sleuth sfdata=data.frame(sample=list.files("results/salmon"), path=sfdirs, condition=samples$condition, stringsAsFactors = F) design = ~condition names(tx2gene)=c("target_id","HGNC") so <- sleuth_prep(sfdata, design, target_mapping = tx2gene,num_cores = 1) # # # Sleuth fit so <- sleuth_fit(so) # # # Extract expression data oe <- sleuth_wt(so, 'conditiontumor') # # # Sleuth results as data frame sleuth_results_oe=sleuth_results(oe, 'conditiontumor', show_all = TRUE) # # # Remove rows with no sloe=sleuth_results_oe[complete.cases(sleuth_results_oe),] write.csv(sloe, "results/sleuth_results/sleuth_expression_results.txt", sep="\t") # # # Merge gene names from tx2gene object and order by qvalue mer_sloe=merge(sloe, tx2gene, all.x=T) mer_sloe[order(mer_sloe$qval),] # # # Write the results to hard disk write.csv(sloe, "results/sleuth_results/sleuth_expression_results_merged.txt", sep="\t") # # # Save the workflow to HDD save.image("results/sleuth_results/sleuth_results.Rdata") |
38 | shell: "rm -rf .snakemake/" |
Support
Do you know this workflow well? If so, you can
request seller status , and start supporting this workflow.
- Share:
-

-

-

Created: 1yr ago
Updated: 1yr ago
Maitainers:
public
URL:
https://github.com/svsuresh/salmon_deseq2_snakemake
Name:
salmon_deseq2_snakemake
Version:
1
Downloaded:
0
Copyright:
Public Domain
License:
None
Keywords:
- Future updates
Related Workflows

ENCODE pipeline for histone marks developed for the psychENCODE project
psychip pipeline is an improved version of the ENCODE pipeline for histone marks developed for the psychENCODE project.
The o...

Near-real time tracking of SARS-CoV-2 in Connecticut
Repository containing scripts to perform near-real time tracking of SARS-CoV-2 in Connecticut using genomic data. This pipeli...

snakemake workflow to run cellranger on a given bucket using gke.
A Snakemake workflow for running cellranger on a given bucket using Google Kubernetes Engine. The usage of this workflow ...

ATLAS - Three commands to start analyzing your metagenome data
Metagenome-atlas is a easy-to-use metagenomic pipeline based on snakemake. It handles all steps from QC, Assembly, Binning, t...
raw sequence reads
Genome assembly
Annotation track
checkm2
gunc
prodigal
snakemake-wrapper-utils
MEGAHIT
Atlas
BBMap
Biopython
BioRuby
Bwa-mem2
cd-hit
CheckM
DAS
Diamond
eggNOG-mapper v2
MetaBAT 2
Minimap2
MMseqs
MultiQC
Pandas
Picard
pyfastx
SAMtools
SemiBin
Snakemake
SPAdes
SqueezeMeta
TADpole
VAMB
CONCOCT
ete3
gtdbtk
h5py
networkx
numpy
plotly
psutil
utils
metagenomics

RNA-seq workflow using STAR and DESeq2
This workflow performs a differential gene expression analysis with STAR and Deseq2. The usage of this workflow is described ...

This Snakemake pipeline implements the GATK best-practices workflow
This Snakemake pipeline implements the GATK best-practices workflow for calling small germline variants. The usage of thi...